|
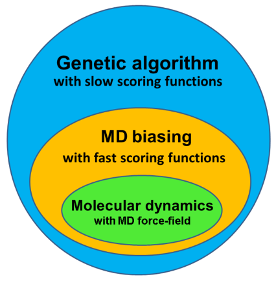
ShiftRefiner is a webserver for refining protein structures with NMR chemical shifts. The method employs biased quenched molecular dynamics, a multi-criterion genetic algorithm, several knowledge-based scoring functions, and structural information derived from NMR chemical shifts.
Reference: Protein Chemical Shift Refinement on the Web, Mark Berjanskii, David Arndt, Noor Hafsa, and David S. Wishart; (Submitted).
|